Ethan's BGGN 213 Portfolio
Class work for bioinformatics
lab09
Ethan Ashley (PID: A15939817) 2025-10-29
- Introduction to PDB
- Visualizing structure data with the Mol* viewer
- Bio3D package for structural bioinformatics
- Play with 3D viewing in R
- Comparative Analysis of Protein Structures
Introduction to PDB
Importing the PDB statistics data file.
#Installing the 'READR' Package
library(readr)
#import data and convert into integer format for numeric operations
PDB_stats <- read_csv("Data Export Summary.csv")
## Rows: 6 Columns: 9
## ── Column specification ────────────────────────────────────────────────────────
## Delimiter: ","
## chr (1): Molecular Type
## dbl (4): Integrative, Multiple methods, Neutron, Other
## num (4): X-ray, EM, NMR, Total
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
#PDB_stats <- read.csv("Data Export Summary.csv", row.names = 1)
#PDB_stats <- lapply(PDB_stats, gsub, pattern = ',', replacement = '')
#PDB_stats <- lapply(PDB_stats, as.integer)
Q1: What percentage of structures in the PDB are solved by X-Ray and Electron Microscopy.
81.4% of the structures in the PDB are solved by X-ray 12.3% of the strucutres in the PDB are solved by electron microscopy.
total_xray <- sum(PDB_stats$`X-ray`)
total_EM <- sum(PDB_stats$EM)
total_entries <- sum(PDB_stats$Total)
total_xray/total_entries
## [1] 0.8143231
total_EM/total_entries
## [1] 0.1227148
Q2: What proportion of structures in the PDB are protein?
86.0% of the structures in the PDB are protein.
PDB_stats$Total[1]/sum(PDB_stats$Total)
## [1] 0.860465
Q3: Type HIV in the PDB website search box on the home page and determine how many HIV-1 protease structures are in the current PDB?
Q3: Make a bar plot overview of Molecular type
library(ggplot2)
ggplot(PDB_stats) + aes(reorder(`Molecular Type`, -Total), Total, fill=PDB_stats$Total) + geom_col()
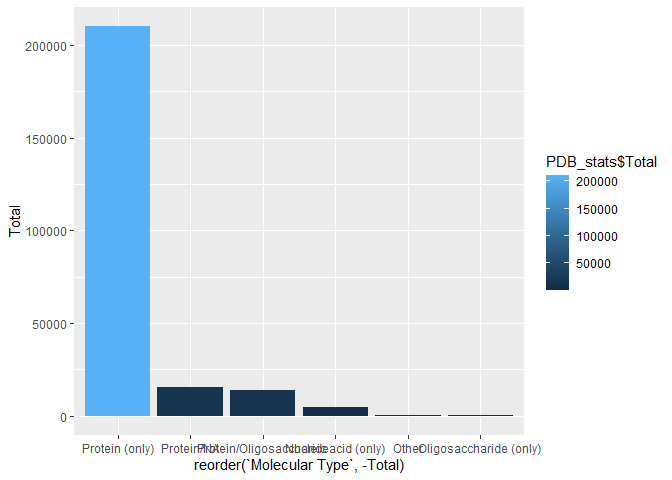
#ggplot(PDB_stats) + aes(reorder(`Molecular Type`, -Total), Total) + geom_bar(stat = "identity", position="stack")
Visualizing structure data with the Mol* viewer
The Mol* viewer is embeded in the website of many bioinformatics websites, like PDB
We will use it on the https://molstar.org/ website
Let’s look at the x-ray crystal structure of the HIV-1 protease bound with the drug indinavir (PDB ID: 1HSG)
Figures or images can be inserted using markdown format.
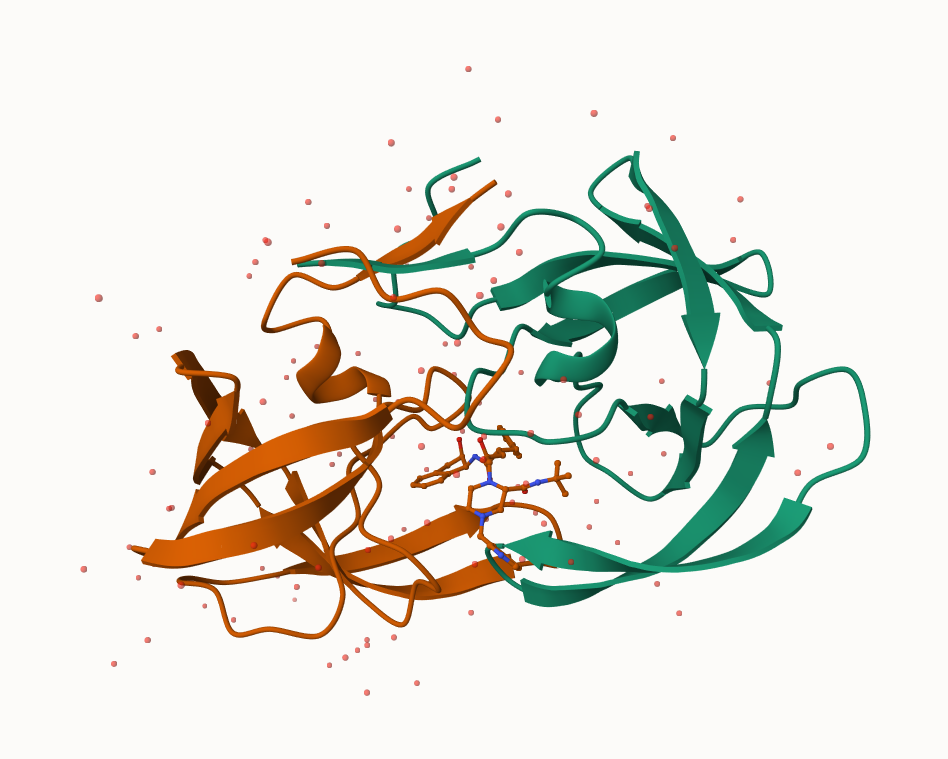
Updated image of the protease with the spacefill model representation of the drug
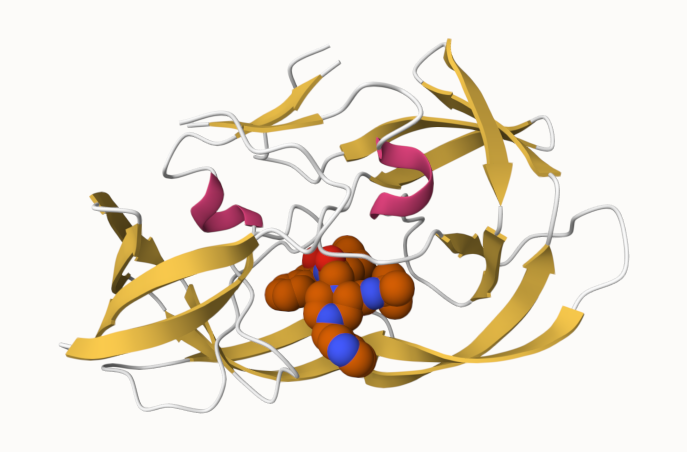
Q4: Water molecules normally have 3 atoms. Why do we see just one atom per water molecule in this structure?
We chose to display the water molecules in a way that is less obtrusive when observing the rest of the structure.
Q5: There is a critical “conserved” water molecule in the binding site. Can you identify this water molecule? What residue number does this water molecule have
This water molecule has residue number 308.
Q6: Generate and save a figure clearly showing the two distinct chains of HIV-protease along with the ligand. You might also consider showing the catalytic residues ASP 25 in each chain and the critical water (we recommend “Ball & Stick” for these side-chains). Add this figure to your Quarto document.
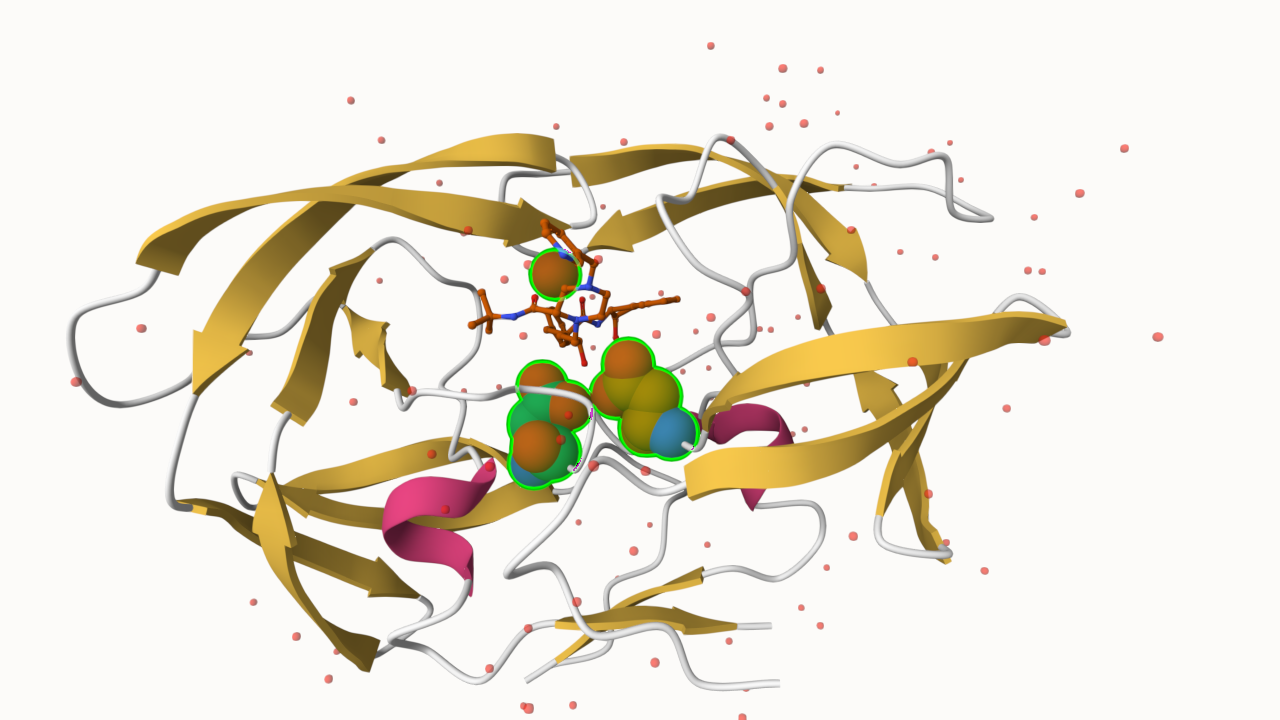
Bio3D package for structural bioinformatics
We can use the bio3d package to read and analyze biomolecular data in R.
library(bio3d)
hiv <- read.pdb("1HSG")
## Note: Accessing on-line PDB file
hiv
##
## Call: read.pdb(file = "1HSG")
##
## Total Models#: 1
## Total Atoms#: 1686, XYZs#: 5058 Chains#: 2 (values: A B)
##
## Protein Atoms#: 1514 (residues/Calpha atoms#: 198)
## Nucleic acid Atoms#: 0 (residues/phosphate atoms#: 0)
##
## Non-protein/nucleic Atoms#: 172 (residues: 128)
## Non-protein/nucleic resid values: [ HOH (127), MK1 (1) ]
##
## Protein sequence:
## PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYD
## QILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFPQITLWQRPLVTIKIGGQLKE
## ALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEICGHKAIGTVLVGPTP
## VNIIGRNLLTQIGCTLNF
##
## + attr: atom, xyz, seqres, helix, sheet,
## calpha, remark, call
Q7: How many amino acid residues are there in this pdb object?
There are 128 residues in this pdb object
Q8: Name one of the two non-protein residues?
HOH
**Q9: How many protein chains are in this structure? **
2, chains A and B.
Let’s get the sequence
pdbseq(hiv)
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
## "P" "Q" "I" "T" "L" "W" "Q" "R" "P" "L" "V" "T" "I" "K" "I" "G" "G" "Q" "L" "K"
## 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40
## "E" "A" "L" "L" "D" "T" "G" "A" "D" "D" "T" "V" "L" "E" "E" "M" "S" "L" "P" "G"
## 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60
## "R" "W" "K" "P" "K" "M" "I" "G" "G" "I" "G" "G" "F" "I" "K" "V" "R" "Q" "Y" "D"
## 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80
## "Q" "I" "L" "I" "E" "I" "C" "G" "H" "K" "A" "I" "G" "T" "V" "L" "V" "G" "P" "T"
## 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 1
## "P" "V" "N" "I" "I" "G" "R" "N" "L" "L" "T" "Q" "I" "G" "C" "T" "L" "N" "F" "P"
## 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
## "Q" "I" "T" "L" "W" "Q" "R" "P" "L" "V" "T" "I" "K" "I" "G" "G" "Q" "L" "K" "E"
## 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41
## "A" "L" "L" "D" "T" "G" "A" "D" "D" "T" "V" "L" "E" "E" "M" "S" "L" "P" "G" "R"
## 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61
## "W" "K" "P" "K" "M" "I" "G" "G" "I" "G" "G" "F" "I" "K" "V" "R" "Q" "Y" "D" "Q"
## 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81
## "I" "L" "I" "E" "I" "C" "G" "H" "K" "A" "I" "G" "T" "V" "L" "V" "G" "P" "T" "P"
## 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99
## "V" "N" "I" "I" "G" "R" "N" "L" "L" "T" "Q" "I" "G" "C" "T" "L" "N" "F"
Let’s trim to chain A and get just it’s sequence:
chainA <- trim.pdb(hiv, chain = "A")
chainA.seq <- pdbseq(chainA)
Let’s blast
#These results will be cached so that it does not re-do the blast search every time
blast <- blast.pdb(chainA.seq)
## Searching ... please wait (updates every 5 seconds) RID = K6TGBU2E014
## ...
## Reporting 249 hits
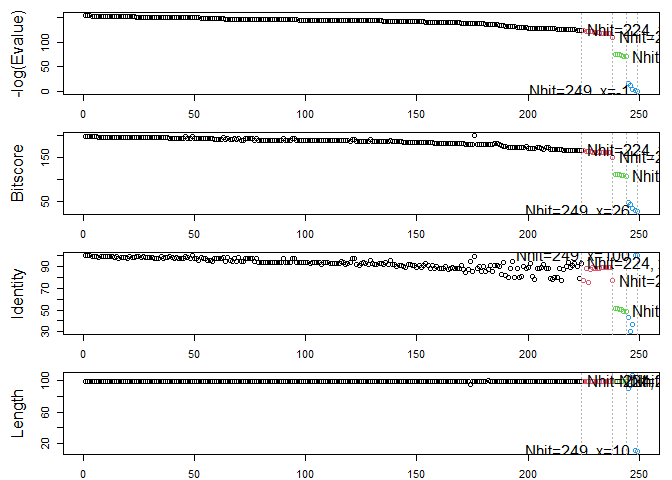
Plot a quick overview of blast results
hits <- plot(blast)
## * Possible cutoff values: 123 110 69 -2
## Yielding Nhits: 224 238 244 249
##
## * Chosen cutoff value of: 69
## Yielding Nhits: 244
 # Prediciton
of functional motions
# Prediciton
of functional motions
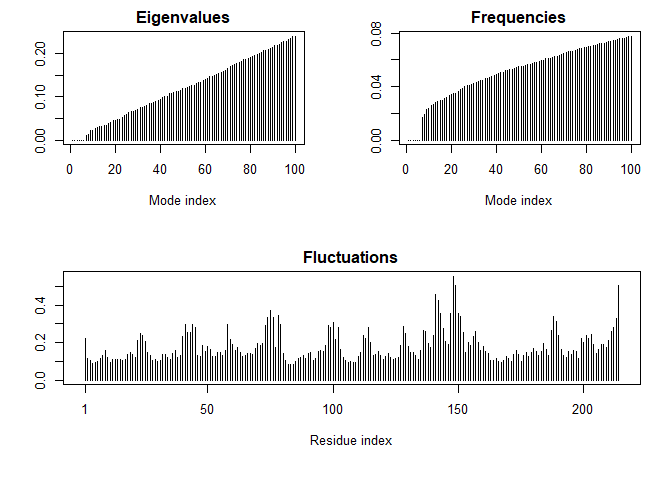
We can run a normal mode analysis (NMA) to predict large scale motions/flexibility/dynamics of any biomolecule that we can read into R.
Let’s look at ADK chain A
adk <- read.pdb("1ake")
## Note: Accessing on-line PDB file
## PDB has ALT records, taking A only, rm.alt=TRUE
adk_A <- trim.pdb(adk, chain="A")
m <- nma(adk_A)
## Building Hessian... Done in 0.03 seconds.
## Diagonalizing Hessian... Done in 0.26 seconds.
plot(m)
Let’s write out a “trajectory” of predicted motion that we can open in Mol*
mktrj(m, file="adk_nma.pdb")
Play with 3D viewing in R
We can use the new bio3dview package, which is not yet on CRAN, to render interactive 3D views in R and HTML output reports.
To install from GitHub we can use the **pak* package
#pak::pak("bioboot/bio3dview")
Comparative Analysis of Protein Structures
Starting with sequence or structure ID (accession number) let’s run a complete analysis pipeline.
library(bio3d)
id <- "1ake_A"
aa <- get.seq(id)
## Warning in get.seq(id): Removing existing file: seqs.fasta
## Fetching... Please wait. Done.
aa
## 1 . . . . . 60
## pdb|1AKE|A MRIILLGAPGAGKGTQAQFIMEKYGIPQISTGDMLRAAVKSGSELGKQAKDIMDAGKLVT
## 1 . . . . . 60
##
## 61 . . . . . 120
## pdb|1AKE|A DELVIALVKERIAQEDCRNGFLLDGFPRTIPQADAMKEAGINVDYVLEFDVPDELIVDRI
## 61 . . . . . 120
##
## 121 . . . . . 180
## pdb|1AKE|A VGRRVHAPSGRVYHVKFNPPKVEGKDDVTGEELTTRKDDQEETVRKRLVEYHQMTAPLIG
## 121 . . . . . 180
##
## 181 . . . 214
## pdb|1AKE|A YYSKEAEAGNTKYAKVDGTKPVAEVRADLEKILG
## 181 . . . 214
##
## Call:
## read.fasta(file = outfile)
##
## Class:
## fasta
##
## Alignment dimensions:
## 1 sequence rows; 214 position columns (214 non-gap, 0 gap)
##
## + attr: id, ali, call
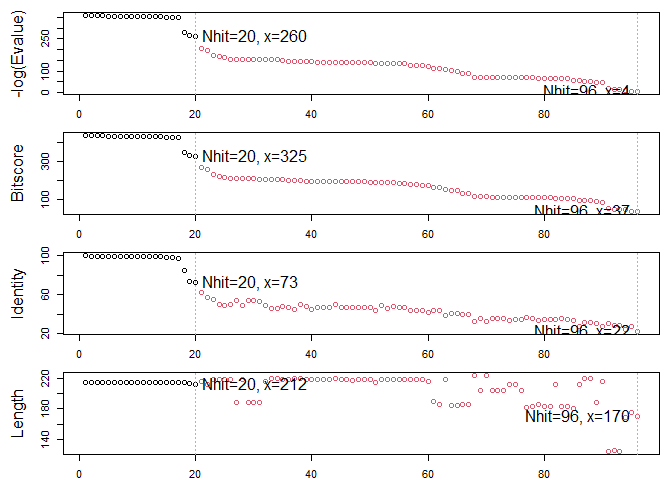
blast <- blast.pdb(aa)
## Searching ... please wait (updates every 5 seconds) RID = K6TGZKDF016
## .
## Reporting 96 hits
hits <- plot(blast)
## * Possible cutoff values: 260 3
## Yielding Nhits: 20 96
##
## * Chosen cutoff value of: 260
## Yielding Nhits: 20
hits$pdb.id
## [1] "1AKE_A" "8BQF_A" "4X8M_A" "6S36_A" "9R6U_A" "9R71_A" "8Q2B_A" "8RJ9_A"
## [9] "6RZE_A" "4X8H_A" "3HPR_A" "1E4V_A" "5EJE_A" "1E4Y_A" "3X2S_A" "6HAP_A"
## [17] "6HAM_A" "8PVW_A" "4K46_A" "4NP6_A"
# Download releated PDB files
files <- get.pdb(hits$pdb.id, path="pdbs", split=TRUE, gzip=TRUE)
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/1AKE.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/8BQF.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/4X8M.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/6S36.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/8Q2B.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/8RJ9.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/6RZE.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/4X8H.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/3HPR.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/1E4V.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/5EJE.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/1E4Y.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/3X2S.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/6HAP.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/6HAM.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/8PVW.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/4K46.pdb exists. Skipping download
## Warning in get.pdb(hits$pdb.id, path = "pdbs", split = TRUE, gzip = TRUE):
## pdbs/4NP6.pdb exists. Skipping download
## | | | 0% | |==== | 5% | |======= | 10% | |========== | 15% | |============== | 20% | |================== | 25% | |===================== | 30% | |======================== | 35% | |============================ | 40% | |================================ | 45% | |=================================== | 50% | |====================================== | 55% | |========================================== | 60% | |============================================== | 65% | |================================================= | 70% | |==================================================== | 75% | |======================================================== | 80% | |============================================================ | 85% | |=============================================================== | 90% | |================================================================== | 95% | |======================================================================| 100%
# Align releated PDBs
#library(BiocManager)
pdbs <- pdbaln(files, fit = TRUE, exefile="msa")
## Reading PDB files:
## pdbs/split_chain/1AKE_A.pdb
## pdbs/split_chain/8BQF_A.pdb
## pdbs/split_chain/4X8M_A.pdb
## pdbs/split_chain/6S36_A.pdb
## pdbs/split_chain/9R6U_A.pdb
## pdbs/split_chain/9R71_A.pdb
## pdbs/split_chain/8Q2B_A.pdb
## pdbs/split_chain/8RJ9_A.pdb
## pdbs/split_chain/6RZE_A.pdb
## pdbs/split_chain/4X8H_A.pdb
## pdbs/split_chain/3HPR_A.pdb
## pdbs/split_chain/1E4V_A.pdb
## pdbs/split_chain/5EJE_A.pdb
## pdbs/split_chain/1E4Y_A.pdb
## pdbs/split_chain/3X2S_A.pdb
## pdbs/split_chain/6HAP_A.pdb
## pdbs/split_chain/6HAM_A.pdb
## pdbs/split_chain/8PVW_A.pdb
## pdbs/split_chain/4K46_A.pdb
## pdbs/split_chain/4NP6_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
## . PDB has ALT records, taking A only, rm.alt=TRUE
## .. PDB has ALT records, taking A only, rm.alt=TRUE
## . PDB has ALT records, taking A only, rm.alt=TRUE
## . PDB has ALT records, taking A only, rm.alt=TRUE
## . PDB has ALT records, taking A only, rm.alt=TRUE
## . PDB has ALT records, taking A only, rm.alt=TRUE
## . PDB has ALT records, taking A only, rm.alt=TRUE
## .. PDB has ALT records, taking A only, rm.alt=TRUE
## .. PDB has ALT records, taking A only, rm.alt=TRUE
## .... PDB has ALT records, taking A only, rm.alt=TRUE
## . PDB has ALT records, taking A only, rm.alt=TRUE
## . PDB has ALT records, taking A only, rm.alt=TRUE
## ..
##
## Extracting sequences
##
## pdb/seq: 1 name: pdbs/split_chain/1AKE_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
## pdb/seq: 2 name: pdbs/split_chain/8BQF_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
## pdb/seq: 3 name: pdbs/split_chain/4X8M_A.pdb
## pdb/seq: 4 name: pdbs/split_chain/6S36_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
## pdb/seq: 5 name: pdbs/split_chain/9R6U_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
## pdb/seq: 6 name: pdbs/split_chain/9R71_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
## pdb/seq: 7 name: pdbs/split_chain/8Q2B_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
## pdb/seq: 8 name: pdbs/split_chain/8RJ9_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
## pdb/seq: 9 name: pdbs/split_chain/6RZE_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
## pdb/seq: 10 name: pdbs/split_chain/4X8H_A.pdb
## pdb/seq: 11 name: pdbs/split_chain/3HPR_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
## pdb/seq: 12 name: pdbs/split_chain/1E4V_A.pdb
## pdb/seq: 13 name: pdbs/split_chain/5EJE_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
## pdb/seq: 14 name: pdbs/split_chain/1E4Y_A.pdb
## pdb/seq: 15 name: pdbs/split_chain/3X2S_A.pdb
## pdb/seq: 16 name: pdbs/split_chain/6HAP_A.pdb
## pdb/seq: 17 name: pdbs/split_chain/6HAM_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
## pdb/seq: 18 name: pdbs/split_chain/8PVW_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
## pdb/seq: 19 name: pdbs/split_chain/4K46_A.pdb
## PDB has ALT records, taking A only, rm.alt=TRUE
## pdb/seq: 20 name: pdbs/split_chain/4NP6_A.pdb
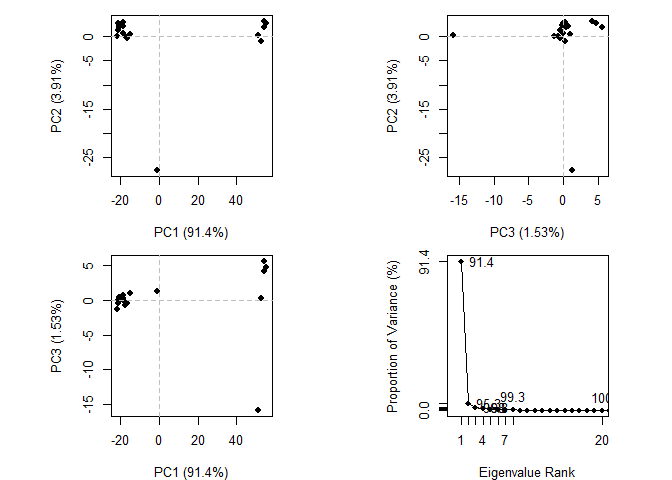
# Perform PCA
pc.xray <- pca(pdbs)
plot(pc.xray)
# Calculate RMSD
rd <- rmsd(pdbs)
## Warning in rmsd(pdbs): No indices provided, using the 182 non NA positions
# Structure-based clustering
hc.rd <- hclust(dist(rd))
grps.rd <- cutree(hc.rd, k=3)
plot(pc.xray, 1:2, col="grey50", bg=grps.rd, pch=21, cex=1)
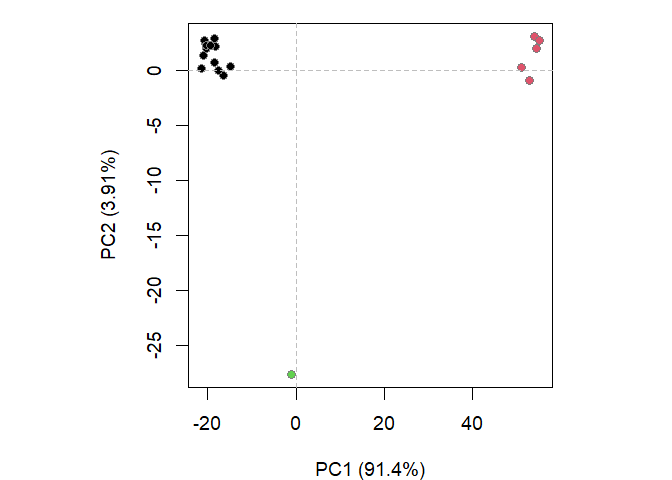
mktrj(pc.xray, file="pca_results.pdb")
library(bio3dview)
#view.pca(pc.xray)